展开查看详情
1 .ASSESSING DRUG SAFETY
USING AI
COMPUTER AIDED DRUG DISCOVERY
AND DEVELOPMENT
�
2 .ABOUT ME
¡ Vishnu Vettrivel - vishnu@wisecube.ai
¡ Data Science/AI platform Architect
¡ NOT a Molecular Biologist or a Medicinal Chemist !
¡ Will be talking about things learnt mostly on the job
¡ Have been working with a Molecular biologist in a
Biotech research firm to help accelerate drug
discovery using Machine learning
�
3 . ¡ History ¡ Computer-aided Drug
¡ Nature as Source
Design
¡ Recent efforts ¡ Molecular Representation
¡ Rational drug discovery ¡ Drug safety assessment
AGENDA ¡ Drug targeting ¡ Demo
¡ Screening ¡ Tools and DBs
¡ Drug Discovery Cycle ¡ Resources
¡ Economics ¡ Summary
�
4 .HISTORY OF
DRUG
DISCOVERY
�
5 . ANCIENT METHODS: NATURE AS A SOURCE
¡ Search for Drugs not new:
¡ Traditional Chinese medicine and Ayurveda both
several thousand years old
¡ Many compounds now being studied
¡ Aspirin’s chemical forefather known to Hippocrates
¡ Even inoculation at least 2000 years old
¡ But also resulted in many ineffective drugs
source: https://amhistory.si.edu/polio/virusvaccine/history.htm
�
6 .MORE RECENT EFFORTS
¡ In 1796, Jenner finds first vaccine: cowpox prevents
smallpox
¡ 1 century later, Pasteur makes vaccines against
anthrax and rabies
¡ Sulfonamides developed for antibacterial purposes
in 1930s
¡ Penicillin: the “miracle drug”
¡ 2nd half of 20th century: use of modern chemical
techniques to create explosion of medicines
�
7 .PROCESS OF FINDING NEW MEDICATIONS MOST COMMONLY AN ORGANIC SMALL INVOLVES THE DESIGN OF MOLECULES
BASED ON THE KNOWLEDGE OF A MOLECULE THAT ACTIVATES OR INHIBITS THAT ARE COMPLEMENTARY IN SHAPE
BIOLOGICAL TARGET. THE FUNCTION OF A PROTEIN AND CHARGE TO THE BIOMOLECULAR
TARGET
RATIONAL DRUG DISCOVERY
�
8 . DRUG TARGET IDENTIFICATION
¡ Different approaches to look for drug targets
¡ Phenotypic screening
¡ gene association studies
¡ chemo proteomics
¡ Transgenetic organisms
¡ Imaging
¡ Biomarkers
Source: https://www.roche.com/research_and_development/drawn_to_science/target_identification.htm
�
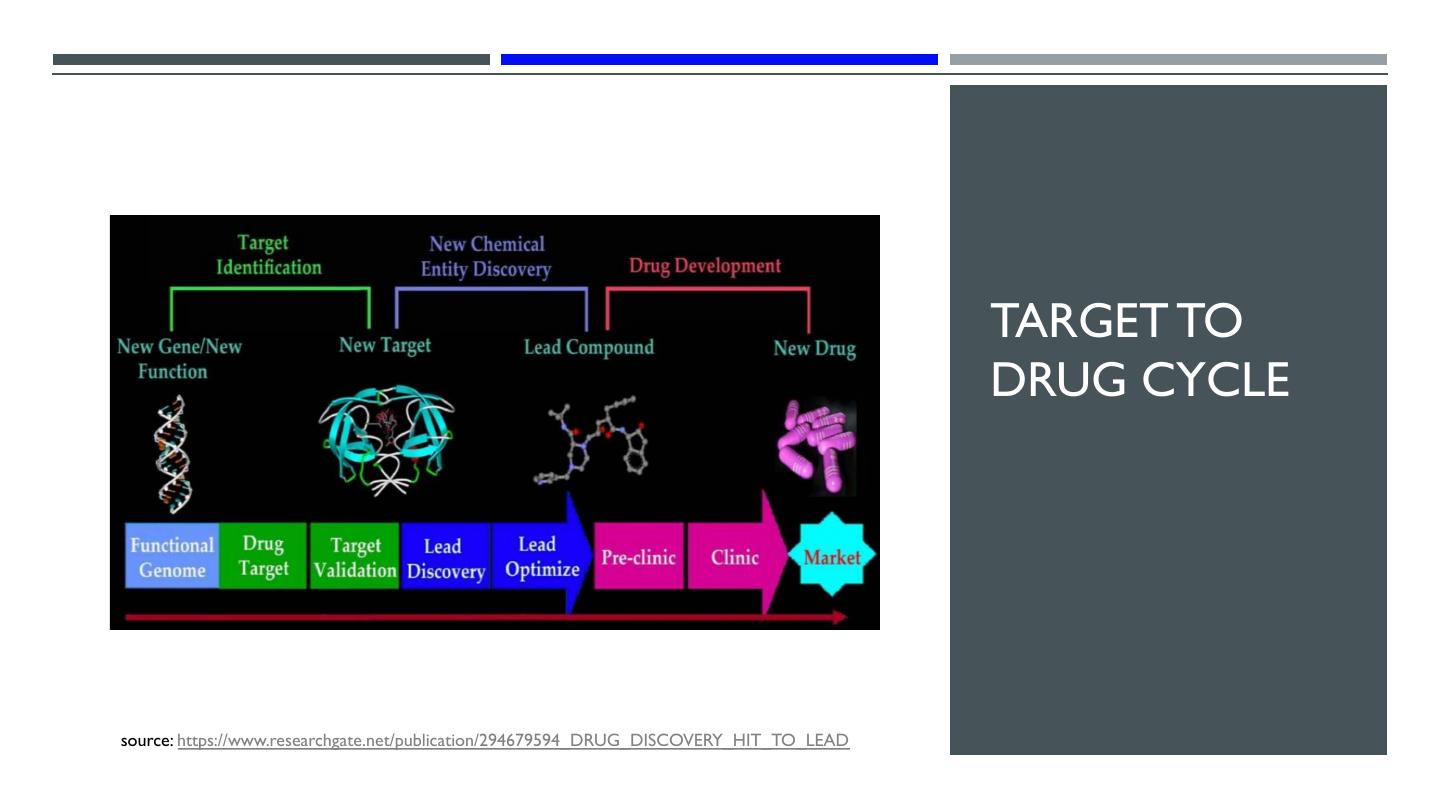
9 . TARGET TO
DRUG CYCLE
source: https://www.researchgate.net/publication/294679594_DRUG_DISCOVERY_HIT_TO_LEAD
�
10 .SCREENING
¡ High Throughput Screening
¡ Implemented in 1990s, still going strong
¡ Allows scientists to test 1000’s of potential targets
¡ Library size is around 1 million compounds
¡ Single screen program cost ~$75,000
¡ Estimated that only 4 small molecules with roots in
combinatorial chemistry made it to clinical
development by 2001
¡ Can make library even bigger if you spend more, but
can’t get comprehensive coverage
¡ Similarity paradox
¡ Slight change can mean difference between active and
inactive
�
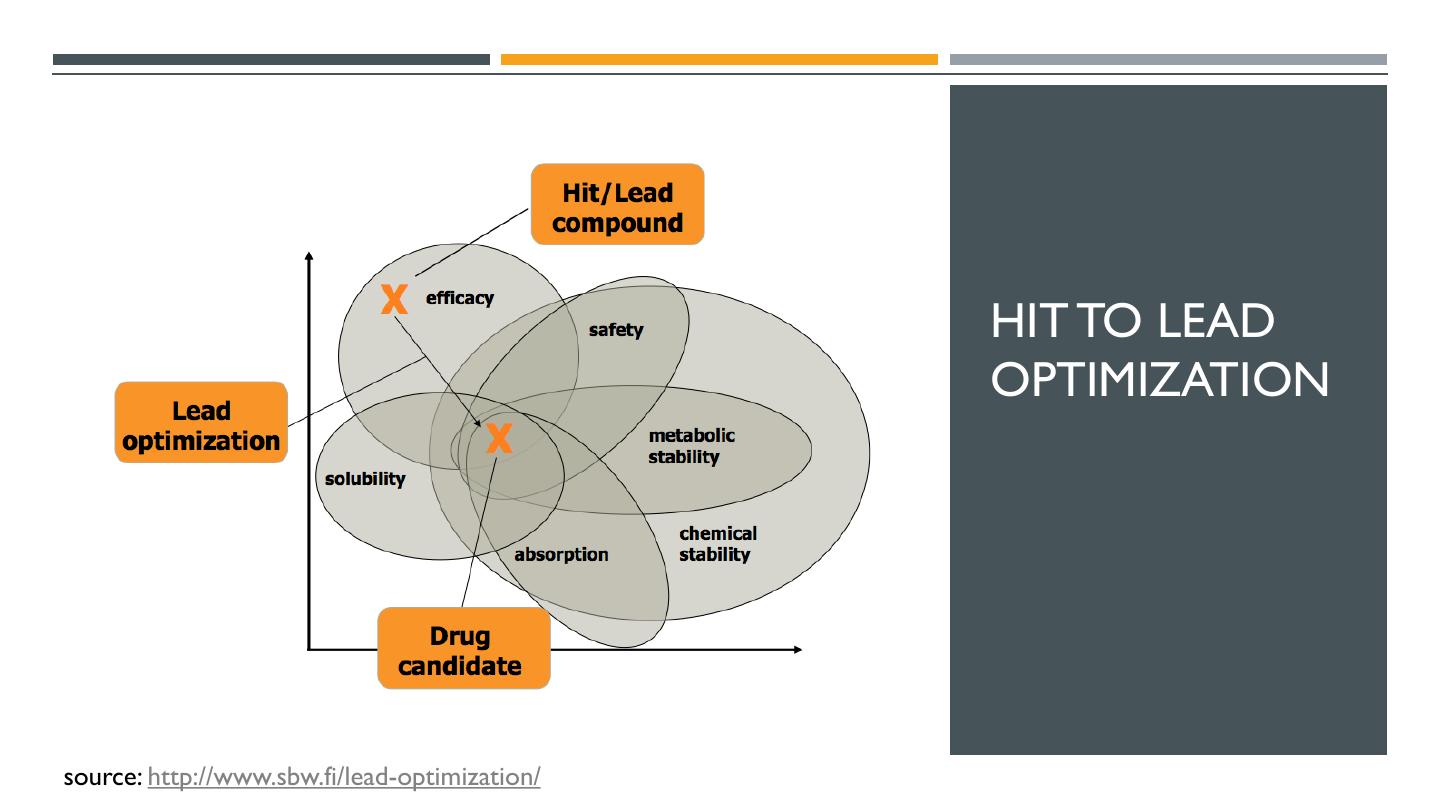
11 . HIT TO LEAD
OPTIMIZATION
source: http://www.sbw.fi/lead-optimization/
�
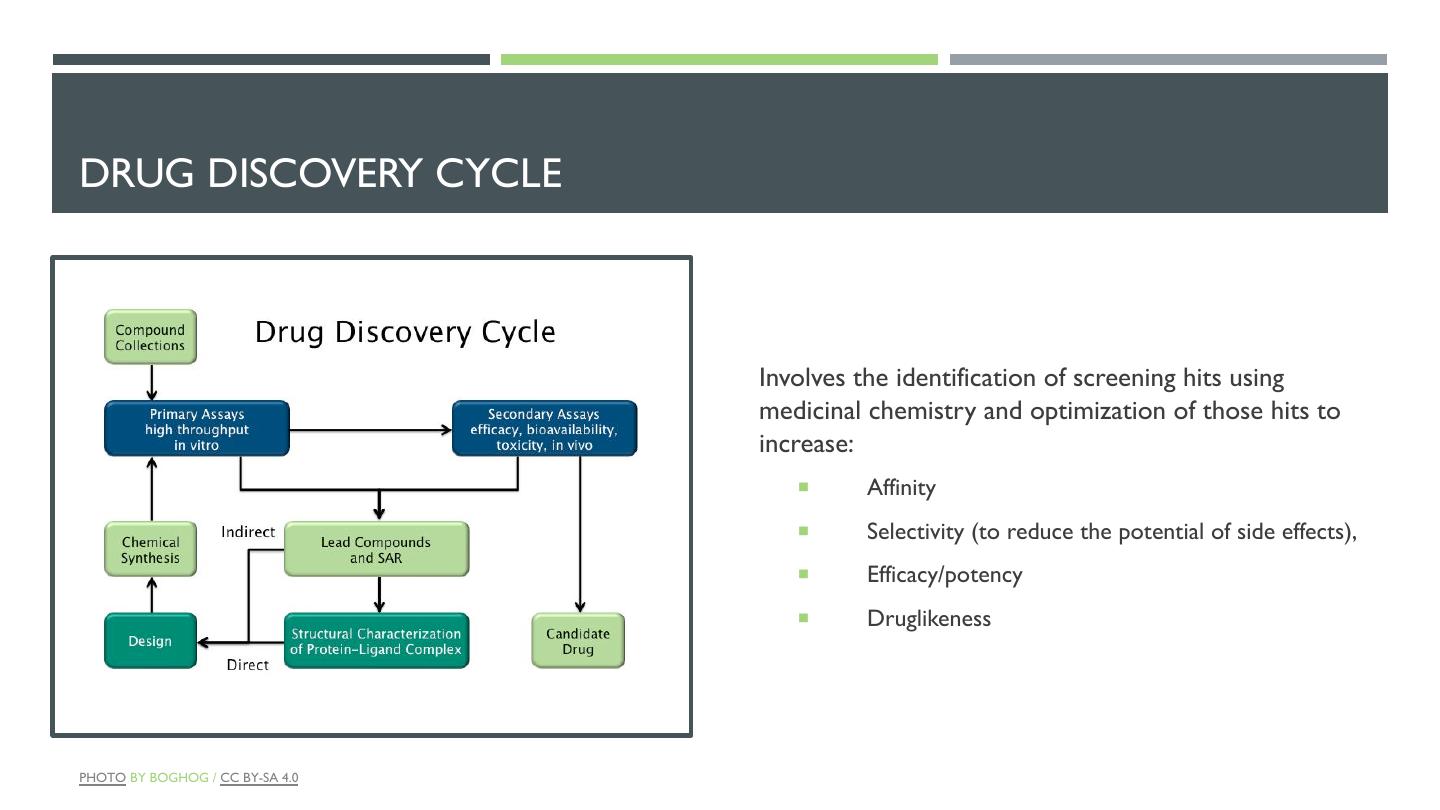
12 .DRUG DISCOVERY CYCLE
Involves the identification of screening hits using
medicinal chemistry and optimization of those hits to
increase:
¡ Affinity
¡ Selectivity (to reduce the potential of side effects),
¡ Efficacy/potency
¡ Druglikeness
PHOTO BY BOGHOG / CC BY-SA 4.0
�
13 .ECONOMICS
Eroom’s Law: Opposite
of Moore’s Law –
Signals worrying trends
in number and cost of
Drugs to Market for
the Pharma industry
source: https://www.nature.com/articles/nrd3681
�
14 .DRUG DISCOVERY
TIMELINE
source: https://www.innoplexus.com/blog/five-reasons-to-embrace-data-driven-drug-development/
�
15 . COMPUTER-
AIDED DRUG
DESIGN
source: http://poster123.info/?u=Pharmacological+Strategies+To+Contend+Against+Myocardial
�
16 .MOLECULAR
REPRESENTATION
�
17 . 1-D DESCRIPTORS
¡ Molecular properties often used for rough
classifications
¡ molecular weight, solubility, charge, number of
rotatable bonds, atom types, topological polar surface
area etc.
¡ Molecular properties like partition coefficient,
or logP, which measures the ratio of solubilities in
two different substances.
¡ The Lipinski rule of 5 is a simple rule of thumb that
is often used to pre-filter drug candidates
Source: chemical Reactivity, Drug-Likeness and Structure Activity/Property Relationship Studies of 2,1,3-Benzoxadiazole Derivatives as Anti-Cancer Activity
�
18 .2-D DESCRIPTORS
¡ A common way of mapping variably structures
molecules into a fixed-size descriptor vector is
“fingerprinting”
¡ circular fingerprints are in more widespread use
today.
¡ A typical size of the bit vector is 1024
¡ The similarity between two molecules can be
estimated using the Tanimoto coefficient
¡ One standard implementation are extended circular
fingerprints (termed ECFPx,with a
number x designating the maximum diameter; e,g,
ECFP4 for a radius of 2 bonds)
�
19 .QSAR
¡ Predictive statistical models correlating one or more
piece of response data about chemicals
¡ Statistical tools, including regression and classification-
based strategies, are used to analyze the response and
chemical data and their relationship
¡ Have been part of scientific study for many years. As
early as 1863, Cros found that the toxicity of alcohols
increased with decreasing aqueous solubility
¡ Machine learning tools are also very effective in
developing predictive models, particularly when
handling high-dimensional and complex chemical data
showing a nonlinear relationship with the responses of
the chemicals
�
20 .SMILE STRING
¡ SMILES (“Simplified molecular-input line-entry system”)
¡ Represents molecules in the form of ASCII character strings
¡ Several equivalent ways to write the same compound
¡ Workaround is to use the canonical version of SMILE
¡ SMILES are reasonably human-readable
�
21 .NEURAL FINGERPRINTS
¡ Hash function can be replaced by a neural network
¡ Final fingerprint vector is the sum over a number of
atom-wise softmax operations
¡ Similar to the pooling operation in standard neural
networks
¡ Can be more smooth than predefined circular
fingerprints
¡ Auto-encoders are also used to find compact latent
representations
¡ converts discrete representations of molecules to and
from a multidimensional continuous representation
�
22 .DRUG SAFETY ASSESSMENT
¡ According to Tufts Center for the Study of Drug Development
(CSDD) the three main causes of failures in Phase III trials:
¡ Efficacy (or rather lack thereof) — i.e., failure to meet the
primary efficacy endpoint
¡ Safety (or lack thereof) — i.e., unexpected adverse or serious
adverse events
¡ Commercial / financial — i.e., failure to demonstrate value
compared to existing therapy
¡ According to another study by Yale School of Medicine
¡ 71 of the 222 drugs approved in the first decade of the
millennium were withdrawn
¡ Took a median of 4.2 years after the drugs were approved for
these safety concerns to come to light
¡ Drugs ushered through the FDA's accelerated approval
process were among those that had higher rates of safety
interventions
�
23 .TOX21 CHALLENGE
¡ Challenge was designed to help scientists
understand the potential of the chemicals and
compounds being tested
¡ The goal was to "crowdsource" data analysis by
independent researchers to reveal how well they
can predict compounds' interference in biochemical
pathways using only chemical structure data.
¡ The computational models produced from the
challenge would become decision-making tools for
government agencies
¡ NCATS provided assay activity data and chemical
structures on the Tox21 collection of ~10,000
compounds (Tox21 10K).
�
24 .DEEPTOX
¡ Normalizes the chemical representations of the compounds
¡ Computes a large number of chemical descriptors that are used as input
to machine learning methods
¡ Trains models, evaluates them, and combines the best of them to
ensembles
¡ Predicts the toxicity of new compounds
¡ Had the highest performance of all computational methods
¡ Outperformed naive Bayes, SVM, and random forests
�
25 .MULTI-TASK
LEARNING
¡ They were able to apply multi-
task learning in the Tox21
challenge because most of the
compounds were labeled for
several tasks
¡ Multi-task learning has been
shown to enhance the
performance of DNNs when
predicting biological activities at
the protein level
¡ Since the twelve different tasks
of the Tox21 challenge data were
highly correlated, they
implemented multi-task learning
in the DeepTox pipeline.
¡
�
26 .ASSOCIATIONS TO
TOXICOPHORES
¡ The histogram (A) shows the
fraction of neurons in a layer that
yield significant correlations to a
toxicophore. With an increasing
level of the layer, the number of
neurons with significant
correlation decreases.
¡ The histogram shows the
number of neurons in a layer
that exceed a correlation
threshold of 0.6 to their best
correlated toxicophore.
Contrary to (A) the number of
neurons increases with the
network layer. Note that each
layer consisted of the same
number of neurons.
�
27 .FEATURE
CONSTRUCTION
BY DEEP
LEARNING.
¡ Neurons that have learned to detect
the presence of toxicophores.
¡ Each row shows a particular hidden
unit in a learned network that
correlates highly with a particular
known toxicophore feature.
¡ The row shows the three chemical
compounds that had the highest
activation for that neuron.
¡ Indicated in red is the toxicophore
structure from the literature that the
neuron correlates with. The first row
and the second row are from the first
hidden layer, the third row is from a
higher-level layer.
�
29 . ¡ Rdkit collection of cheminformatics and machine-learning
software written in C++ and Python.
¡ DeepChem is an integrated python library for chemistry and
drug discovery; it comes with a collection of implementations
for many deep learning based algorithms.
¡ Chembl is a public database containing millions of bioactive
molecules and assay results. The data has been manually
TOOLS AND transcribed and curated from publications. Chembl is an
invaluable source, but has its share of errors—e.g., sometimes
affinities are off by exactly 3 or 6 orders of magnitude due to
DATABASES wrongly transcribed units (micromols instead of nanomols).
¡ PDBbind is another frequently used database, which contains
protein-ligand co-crystal structures together with binding
affinity values. Again, while certainly very valuable, PDBbind has
some well-known data problems.
¡ https://www.click2drug.org/ website containing a comprehensive
list of computer-aided drug design (CADD) software, databases
and web services.
�